
As questões ligadas ao rol de procedimentos e eventos em saúde já passaram por diversos capítulos na história recente e infelizmente não há previsão, ao menos a curto prazo, para o fim das disputas envolvendo a regulação da extensão de coberturas obrigatórias dos contratos de planos de saúde.
O mais recente capítulo dessa história, ainda em fase inicial e caminhando silenciosamente, é a ação judicial promovida pela SINDUSFARMA em face da ANS, dada a questão ligada a regulação dos produtos imunobiológicos.
A questão é complexa e faremos uma apresentação breve da demanda considerando nossa posição em prol dos atos regulatórios da ANS, apresentaremos pontos que entendemos sensíveis sobre a questão do ponto de vista jurídico.
A questão pode ser abordada por diversos aspectos sociais, políticos e econômicos, mas, nossa opção é pelo viés jurídico nos atendo a questões jurídicas ligadas ao tema de forma apenas superficial e indicativa sobre a questão.
O intuito da opinião formalizada neste artigo é a contribuição com o debate institucional sobre o tema e fomentando o debate da política regulatória sobre o rol ligada a este tema que é extremamente relevante para o setor.
Em resumo, a ação aborda uma ação anulatória de ato administrativo da Agência Nacional de Saúde Suplementar (ANS), especificamente relacionada à decisão tomada na Reunião Ordinária nº 594, realizada em 04 de setembro de 2023. A deliberação em questão foi baseada na Nota Técnica 03/2023 da ANS, visando excluir os “produtos de terapia avançada” da cobertura automática por planos e operadoras de saúde.
A SINDUSFARMA questiona a validade da deliberação da Diretoria Colegiada da ANS, argumentando que não foram seguidos os procedimentos administrativos típicos, conforme estabelecido no Regimento Interno da Agência, especificamente na Resolução Regimental nº 21/2022. A principal controvérsia reside no fato de a decisão ter sido registrada apenas na Ata da Reunião nº 594/2023, sem a adoção de atos administrativos formais.
Além disso, a ação alega que a deliberação da ANS é juridicamente inválida por diversos motivos. Entre eles, destacam-se vícios de formalidade e forma, por não cumprir os procedimentos estabelecidos no regimento interno da Agência. Ademais, a decisão entra em conflito com deliberações da Agência Nacional de Vigilância Sanitária (ANVISA), que classifica os “produtos de terapia avançada” como medicamentos. Também é apontada a contrariedade da deliberação da ANS com as disposições da Lei nº 9.656/1998, Lei nº 14.307/2022 e Lei nº 14.454/2022.
Esses argumentos formam a base da ação anulatória, buscando invalidar a decisão da ANS e garantir a cobertura securitária automática dos “produtos de terapia avançada” pelos planos e operadoras de saúde.
O centro do requerimento do SINDUSFARMA está em garantir a incorporação automática do chamado Produto de Terapia Avançada no rol de procedimentos e eventos em saúde da ANS.
A primeira questão a ser avaliada é o conceito técnico do produto de terapia avançada. Segundo a ANVISA[1]
Os produtos de terapia avançada são produtos biológicos, utilizados com fins terapêuticos, obtidos a partir de células e tecidos humanos que foram submetidos a um processo de fabricação; ou produtos que consistem em ácidos nucleicos recombinantes e que tem como objetivo regular, reparar, substituir, adicionar ou deletar uma sequência genética ou modificar a expressão de um gene.
A regulação destes produtos se deu naquela agência por meio da Resolução de Diretoria Colegiada (RDC) 508/2021, que dispõe sobre a adoção de boas práticas em células humanas para uso terapêutico e pesquisa clínica, pela RDC 506/20, que estabelece regras para a realização de ensaios clínicos com produto de terapia avançada investigacional no Brasil, e pela RDC 505/2021, que dispõe sobre o registro de produto de terapia avançada.
Do ponto de vista técnico, estamos diante de produto/medicamento excepcional. Atualmente encontram-se registrados 05 (cinco) produtos[2]: Carvykti, kymriah, luxturna, yescarta e o Zolgensma.
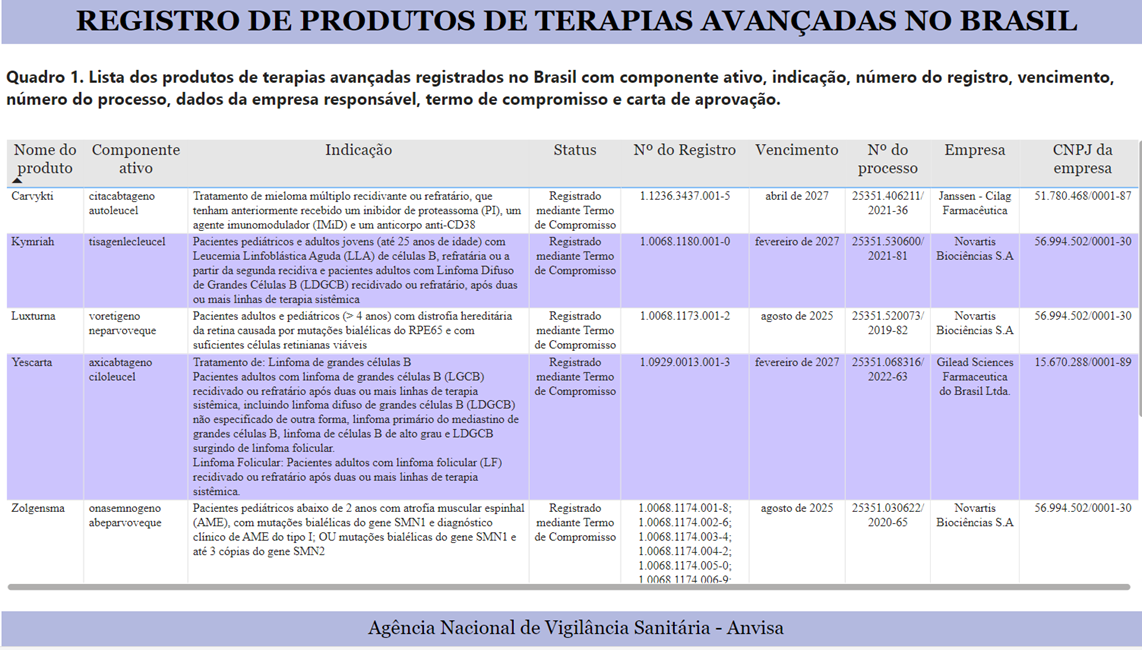
Corrobora a excepcionalidade a Nota Técnica nº 3/2023/GCITS/GGRAS/DIRAD-DIPRO/DIPRO/ANS de onde extraímos:
G) “A complexidade e a singularidade destas terapias avançadas e seus respectivos ‘medicamentos especiais’ pode ser também invocada pelo fato de estarem sob gestão da Gerência de Sangue, Tecidos, Células, Órgãos e Produtos de Terapias Avançadas (GSTCO), e não no âmbito da Gerência-Geral de Medicamentos da ANVISA.”
H) “Corrobora, ainda, a especificidade de tais terapias a criação de uma rede específica de especialistas para apoiar as decisões e estudos em relação ao tema. A Rede Nacional de Especialistas em Terapia Avançada (RENETA), tem por objetivo auxiliar a ANVISA na avaliação de dossiês de ensaios clínicos e registro dos produtos, bem como em processos de monitoramento pós-comercialização dos produtos de terapias avançadas, além de prover treinamento técnico para os especialistas da ANVISA, de acordo com os normativos sanitários brasileiros vigentes e documentos técnicos oficiais nacionais e de autoridade sanitária internacional oficialmente reconhecida, contribuindo de forma decisiva para a transferência de conhecimentos tecnológicos e regulatórios para garantir sustentabilidade na avaliação de segurança, qualidade e eficácia dos produtos de terapias avançadas na assistência à saúde.”
I) “Outro aspecto a diferenciar os medicamentos especiais em Terapia Avançada é a aprovação do registro em caráter condicional trazendo uma série de compromissos por parte do detentor da tecnologia.”
Analisado o conceito técnico de forma objetiva e sua excepcionalidade, conforme dados técnicos da ANS e da ANVISA, a primeira questão analisada está na classificação destes como medicamento. Neste sentido, a RDC 505/21 registra que se trata de produtos de terapia avançada[3], tratando do mesmo como categoria especial de medicamentos novos[4]. Pela simples leitura da RDC e da definição regulatória, fica claro que a ANVISA compreende estes produtos como excepcionais, inseguros e que demandam constante monitoramento.
Tamanha excepcionalidade também está presente na nota técnica nº 4/2023/SEI/GSTCO/GGBIO/DIRE2/ANVISA que em resposta a questionamento da ABRAMGE, registrou a excepcionalidade e controle de produto de terapia avançada registrado.
Com estas considerações iniciais, compreendemos ser possível avançar para os elementos de direito que demonstram a regularidade do cuidado da ANS em classificar tais produtos de forma excepcional.
O centro do mérito está na coordenação regulatória
Diferentemente do argumento presente nas petições iniciais, não há sobreposição regulatória, mas sim, verdadeira coordenação regulatória entre a ANVISA e a ANS.
Inicialmente é necessário registrar que a regulação não possui única ligação com as agências, ao contrário, ela permeia toda a atividade política e legislativa, sendo inerente a atividade das agências dada a sua especificidade. No entanto, é preciso que se compreenda que regular vai além das normas emitidas por agências, veja-se como exemplo todo o imbróglio legislativo presente nas Leis 14.307/22 e 14.454/22, ambas que alteram o Art. 10 da Lei 9.656/98 fixando normas regulatórias sobre incorporação e excepcionalidades sobre o rol de procedimentos e eventos em saúde da ANS.
No que concerne especificamente as agências, o poder regulatório destas emana diretamente do Art. 174 da Constituição[5]. Com a previsão constitucional, foi criada a ANVISA por meio da Lei 9.782/99 e a ANS pela Lei 99.61/00.
Nos termos do previsto no Art. 7º e 8º da Lei 9.782/99 compete a ANVISA especificamente sobre o tema em estudo.
Art. 7º Compete à Agência proceder à implementação e à execução do disposto nos incisos II a VII do art. 2º desta Lei, devendo:
(…)
VII – autorizar o funcionamento de empresas de fabricação, distribuição e importação dos produtos mencionados no art. 8o desta Lei e de comercialização de medicamentos;
VIII – anuir com a importação e exportação dos produtos mencionados no art. 8º desta Lei;
IX – conceder registros de produtos, segundo as normas de sua área de atuação;
(…)
Art. 8º Incumbe à Agência, respeitada a legislação em vigor, regulamentar, controlar e fiscalizar os produtos e serviços que envolvam risco à saúde pública.
§ 1º Consideram-se bens e produtos submetidos ao controle e fiscalização sanitária pela Agência:
I – medicamentos de uso humano, suas substâncias ativas e demais insumos, processos e tecnologias;
(…)
II – imunobiológicos e suas substâncias ativas, sangue e hemoderivados;
Já a ANS, por força do mandamento Legal previsto no Art. 10, §4º da Lei 9.656/98[6], nos termos do Art. 3º e 4º da Lei 9.961/00 tem competência:
Art. 3oA ANS terá por finalidade institucional promover a defesa do interesse público na assistência suplementar à saúde, regulando as operadoras setoriais, inclusive quanto às suas relações com prestadores e consumidores, contribuindo para o desenvolvimento das ações de saúde no País.
(…)
III – elaborar o rol de procedimentos e eventos em saúde, que constituirão referência básica para os fins do disposto na Lei no 9.656, de 3 de junho de 1998, e suas excepcionalidades;
Da leitura da Lei que institui as duas agências identificamos que não há sobreposição regulatória entre as agências, ao contrário, os espaços regulatórios estão bem delimitados cabendo a ANVISA a avaliação de novos produtos e tecnologias para a circulação no território nacional, e a ANS a incorporação em lista de cobertura obrigatória pelas Operadoras de planos privados a saúde.
Aliás, esta é uma distinção clara, porém absolutamente necessária, na medida em que as iniciais da indústria farmacêutica propositalmente “misturam os conceitos”, desconsiderando a distinção entre as atividades das agências. Note que uma tecnologia aprovada pela ANVISA para uso no território nacional, não será incorporada automaticamente no rol da ANS, nem tampouco no PCDT/SUS pelo CONITEC. Ambas as agências (ANS e CONITEC) possuem processos administrativos de incorporação de novas tecnologias, amplamente discutidos e validados.
Identificadas as competências legais das agências, do ponto de vista da estratégia regulatória de ambas as agências estamos diante de normas regulatórias de comando e controle, portanto, as normas regulatórias fixam condições para a incorporação da tecnologia[7]. E isto se justifica porque estamos diante de produtos excepcionais e que demandam além de elevadíssimo controle pela agência de controle sanitário, ainda, a efetiva avaliação técnica por meio de ATS, conforme termos da RN 555/22[8], para incorporação no rol da ANS.
Com todos os elementos jurídico-regulatórios já oportunizados compreendemos que a questão em discussão está ligada a coordenação regulatória entre agências, esta que representa um desafio convencional em todas as agências no mundo, e isto porque, determinados temas são fragmentados em agências distintas, fato este já observado pela literatura especializada[9] que define o fenômeno como[10]:
related jurisdictional assignments, where Congress assigns closely related but distinct roles to numerous agencies in a larger regulatory or administrative regime.
A questão fica clara quando observada pelo próprio tema em análise. Note que cada agência possui competências distintas sobre o mesmo produto de terapia avançada, enquanto a ANVISA buscou meios de excepcionalmente enquadrar tais produtos em uma classe não prevista pela própria agência, a fim de assegurar seu curso no território nacional, e a ANS, diante da excepcionalidade dos mesmos compreendeu pela adoção de medidas regulatórias de controle excepcionais, em linha com a própria regulação ANVISA. Não há nesta hipótese disputas regulatórias entre agências ou mesmo delegação regulatória de poderes equivalentes para ambas, cada agência possui espaços claros de regulação, o que vem sendo observado especialmente pela ANS para a regulação dos produtos de terapia avançada[11].
O comportamento regulatório da ANS é claro: se a ANVISA trata tais produtos como excepcionais, diante das incertezas e riscos, a ANS de forma absolutamente coordenada, adota a mesma postura compreendendo os riscos a saúde dos beneficiários, e ainda, a relação econômica necessária para avaliação para incorporação na saúde privada.
Corrobora esta posição a própria procuradoria da ANS no parecer n. 00019/2023/GECOS/PFANS/PGF/AGU, que apesar de adotar outro caminho, em suas conclusões empreende a mesma análise regulatória de exceção quanto aos produtos de terapia avançada ao afirmar:
A) Tendo em vista a “diversidade, peculiaridade e natureza dos processos envolvidos”, alta complexidade, inovação disruptiva, “riscos e incertezas clínicas”, “perspectiva de falhas de assimetria de informações entre pacientes/cuidadores e o setor produtivo”, “externalidades negativas inerentes”, “dados clínicos inconclusivos quanto à eficácia de seu uso”, “concessão de registros convencionais condicionados à assinatura de Termo de Compromisso pela empresa titular do produto”, e “registro de produtos com necessidade de dados adicionais para comprovação de eficácia clínica”, pode-se afirmar que há motivo sério para excluir os “produtos de terapias avançadas” do alcance do termo “medicamento” previsto no art. 8º, III, da Resolução Normativa – RN nº 465/2021, e no art. 12, II, “d”, da Lei nº 9.656/1998.
B) Não se trata simplesmente de medicamentos novos, mas de uma categoria especial de medicamentos novos, para a qual tem-se entendido ser inviável a aplicação das regras previstas para medicamentos comuns, a não ser supletivamente.
C) Os fins visados pela ANVISA com as regras próprias para registro de produtos de terapias avançadas seriam reforçados, no campo da saúde suplementar, com a submissão desses produtos ao processo de atualização do Rol, considerando as diretrizes que nele são observadas, descritas no art. 3º da RN ANS nº 555, de 14 de dezembro de 2022, que dispõe sobre o rito processual de atualização do Rol de Procedimentos e Eventos em Saúde, ou com sua submissão ao processo administrativo de incorporação de tecnologias em saúde pelo SUS, considerando as diretrizes da CONITEC, descritas no art. 3º do Decreto nº 7.646, de 2011.
Em resumo, a coordenação regulatória está clara: a ANVISA promoveu a incorporação de produto de terapia avançada, criando uma classe excepcional para a sua incorporação que demanda a observação de diversas questões técnicas e de monitoramento. De outro lado, a ANS, identificando que estes produtos foram tratados de forma distinta daquela convencionalmente adotada pela ANVISA, promoveu os atos regulatórios necessários a distinguir estes dos medicamentos comuns, dada a distinção criada pela própria ANVISA, adotando os cuidados necessários na saúde suplementar para alcançar os objetivos próprios da política pública de saúde iniciada pela ANVISA quanto limitação e excepcionalidade destes produtos.
O §13º do Art. 10 da Lei 9.656/98, a autonomia do médico e do paciente
Apesar de parecer claro que há obrigatória necessidade de observância a regulação para o acesso a estes produtos de exceção, o principal argumento da Interfarma tem ligação com a alteração legislativa que cria o §13º do Art. 10 da Lei, compreendendo ainda que a simples autorização pela ANVISA, e a ausência de substituto terapêutico são suficientes para a incorporação automática pela ANS.
Apesar do argumento é necessário registrar que o rol de procedimentos e eventos em saúde da ANS segue vigendo, conforme termos do Art. 10 §4º da Lei, representando segurança aos pacientes, especialmente estes mais sensibilizados pela ocorrência de doenças graves.
Aqui igualmente reside outro ponto sensível para discussão jurídica, a compatibilização entre: as normas regulatórias x o §13º do Art. 10 da Lei x Código de Proteção e Defesa do Consumidor. E isto porque, a maioria das decisões judiciais que asseguram coberturas extra-rol tem como fundamento, além do direito fundamental a vida prevista no Art. 5º da CRFB/88, a alegação da imposição de cláusula abusiva para acesso a terapia que o beneficiário prescinde, o que representaria uma limitação contratual ilícita.
Impõe-se contra este argumento as questões técnicas oportunizadas nos itens anteriores, e ainda, a compreensão sobre os limites da autonomia do médico e do paciente, haja visto que deveria ser este o elemento que modula a limitação de acesso e escolha sobre terapias não chanceladas pelo rol.
Muito brevemente, autonomia pode ser definida como a capacidade de governar-se pelos próprios meios. Para a sua consecução, é necessário que o homem seja capaz de determinar sua vontade pela lei moral, tornando-se consciente de sua liberdade, somente neste contexto de liberdade é possível que o homem proceda de com sua autodeterminação para a uma ação moral[12]. No que toca a autonomia dos pacientes, a bioética largamente estuda o tema e registra que esta se pauta pela trindade bioética, ou seja, pela necessária observância dos princípios da autonomia, beneficência e justiça, inexistindo hierarquia entre estes. Neste contexto, a autonomia do paciente deve ser considerada como basilar da conduta ética, cabendo o respeito a decisão do paciente que deve discernir sobre a decisão de seu tratamento de forma livre e consciente, portanto respeitar a autonomia do paciente é, antes de mais nada, reconhecer sua dignidade humana[13].
Apesar do agir de forma autônoma, todos são vulneráveis, ou seja, estão sujeitos a ofensas físicas ou psicológicas, de forma ou não intencional, por agentes de qualquer natureza, inclusive por acidente. Necessário portanto compreender que o ser humano sempre é vulnerável, o que não se confunde com o estar vulnerável, mas trata-se de uma situação latente, uma possibilidade.[14] Neste contexto, o ser humano em estado de vulnerabilidade vê sua autonomia maculada, seja porquanto perde a capacidade de discernir ou mesmo de decidir em razão do estado de necessidade, ou mesmo, pela ausência de informações adequadas, que somadas a vulnerabilidade maculam o agir com autonomia.
Aliás, estas diretrizes encontram maior relevância quando o paciente está diante de sua terminalidade. E isto porque, aqui para além dos princípios bioéticos estamos diante do flamulo maior ao respeito da dignidade da pessoa humana, atingindo este seu pleno significado[15]. O paciente terminal encontra seu maior momento de vulnerabilidade, e o exercício de sua autonomia apesar de parecer maculado, deve ser plenamente exercido, porquanto suas escolhas sobre a terminalidade da vida devem balizar a atuação das equipes de saúde.
No que concerne a autonomia médica, esta encontra respaldo legal na Lei 12.842/13, que dispõe sobre o exercício da medicina, na Resolução CFM nº 1.931/09 que regulamenta o Código de Ética Médica, bem como nas demais resoluções exaradas pelo CFM. a evolução das ciências médicas e da forma como a informação é difundida, atraem ao médico o dever de também revisar suas escolhas, diante de novas evidências científicas, ou mesmo, pela atuação em colegiado profissional, fatos e elementos estes que limitam a autonomia, atendendo, por conseguinte, a tríade que baliza a bioética.
Importante registrar que não estamos extinguindo a capacidade do médico ou do paciente em agir com autonomia, mas sim, expondo a existência de uma limitação objetiva e clara. Que no caso em vertente se corrobora pela própria criação pela ANVISA da Rede Nacional de Especialistas em Terapia Avançada (RENETA). Note que estes elementos são compatíveis com o próprio marco legal do §13º do Art. 10 da Lei, na medida em que há previsão regulatória para um ciclo mínimo de verificação de cobertura, baseado em programa da ANVISA e da ANS, não havendo que se falar em incorporação automática, e por conseguinte, nem mesmo em lesão a direitos do consumidor, na medida em que cabe as agências promover a segurança do paciente no acesso a produtos médicos, especialmente, este de aprovação excepcionalíssima.
Situação esta já observada no direito brasileiro quando do julgamento da ADI 5.501 que discutia a constitucionalidade da Lei 13.269/16 que regulava a autorização para comercialização da fosfoetalonamina, substância que se dizia eficaz para o tratamento do câncer. Naquela oportunidade, o julgado compreendeu pela inconstitucionalidade da lei, como fundamento, o Min. Relator Marco Aurelio Melo registrou que O direito à saúde não será plenamente concretizado se o Estado deixar de cumprir a obrigação de assegurar a qualidade de droga mediante rigoroso crivo científico, apto a afastar desengano, charlatanismo e efeito prejudicial. E neste contexto defendeu a necessidade de deferência a ANVISA para a autorização de circulação de medicamentos no Brasil.
A Falta de Análise de Impacto Regulatório: Todas as disposições de todas as agências deverão ser declaradas nulas.
O último dos argumentos jurídicos presente na inicial seria a falta de AIR para a tomada de decisão da DICOL, o que inclusive, feriria o Regimento Interno da ANS.
Sobre o tema, compreendemos por poucas considerações. E isto porque, a ANS adotou a medida de forma excepcional diante da incorporação pela ANVISA dos produtos de terapia avançada, a medida em absolutamente nada viola a Lei das agências, ou mesmo, o regulamento da ANS que poderá adotar a medida AIR a qualquer tempo, especialmente diante da controvérsia instalada na demanda.
Adicione-se que não há qualquer prejuízo a incorporação das tecnologias discutidas, veja-se que o Zolgensma já foi incorporado com DUT, e os demais devem ser inscritos no FormRol para que passem pelo processo administrativo previsto pela RN 555/22, meio este definido como adequado pela ANS para a incorporação destas terapias.
Deve ser ainda esclarecido que se o indicado argumento fosse vencedor, ou seja, da falta de AIR ou mesmo da ARR, todas as agências reguladoras do país teriam atos regulatórios declarados nulos, haja visto a própria falta de cultura das agências em promover o AIR e o ARR de forma contínua, argumento este que não poderia ser acatado pelo judiciário sob pena de se criar um cenário de insegurança jurídica determinante para todas as agências do país. No pior cenário, deve o judiciário determinar a realização do AIR conforme o previsto na lei e nas disposições regulatórias da agência.
Não intervenção do judiciário no mérito administrativo
Todo o arcabouço jurídico produzido aponta para uma decisão técnica formulada pela ANS dentro dos espaços regulatórios autorizados pela Lei, e a importância disto para a demanda está no princípio da deferência, pelo qual, o Poder Judiciário não deve interferir no mérito administrativo da Agência que é a detentora do poder regulatório para a tomada de decisão sobre a política pública posta em discussão.
A defesa do ato regulatório da ANS deve considerar a existência de juridicidade em sua escolha, o que é claramente observado em todo o produzido até aqui, especialmente diante da relevância do tema e de seus desdobramentos.
Nesse contexto, a conclusão de eventual demanda deve considerar que se a ANS é competente para a tomada de decisão sobre a política pública, ela o faz de forma a atender ao interesse público, dentro do espaço regulatório que lhe é concedido, não cabe ao Poder Judiciário alterar a decisão da agência (escolhendo por ela) sob pena de violar o princípio da separação dos poderes.
Note que este é um argumento de conclusão, ou seja, analisada a regularidade do ato regulatório e a tomada de decisão de forma a atender a juridicidade e a reflexividade administrativa, fica o P. Judiciário impedido de decidir alterando o mérito da escolha regulatória da ANS.
Concluindo
Nada no tema em estudo é simples. Além das questões jurídicas apresentadas, existem outras ligadas a política regulatória que precisam ser analisadas em conjunto pelo próprio benefício dos consumidores e da sustentabilidade do mercado de Saúde Suplementar.
Em nossa análise, a incorporação sem o devido controle regulatório e até de preços destes produtos imunobiológicos impõe verdadeiro risco a população, dada a necessidade de atenção rigorosa a protocolo de saúde dirigidos ao uso destes produtos, considerando o Brasil extremamente frágil a incorporação e recepção de medicamentos sem a observância de todo o arcabouço regulatório, bem como chance de colapso do sistema de saúde suplementar pela ausência de critérios ligados ao custo destas medicações, considerando que a tabela CEMED faz distinção entre o SUS e o mercado privado.
Como o dito nas primeiras linhas a questão é complexa e nem de longe se pretende exaurir a discussão em torno do tema, que deverá em algum momento ter a devida atenção.
[1] https://www.gov.br/anvisa/pt-br/assuntos/sangue/terapias-avancadas
[2] https://www.gov.br/anvisa/pt-br/assuntos/sangue/terapias-avancadas/produtos-registrados
[3] Vide Arts. 1º e 2º
[4] Vide Art. 4º, inc. XVIII
[5] Art. 174. Como agente normativo e regulador da atividade econômica, o Estado exercerá, na forma da lei, as funções de fiscalização, incentivo e planejamento, sendo este determinante para o setor público e indicativo para o setor privado
[6] § 4º A amplitude das coberturas no âmbito da saúde suplementar, inclusive de transplantes e de procedimentos de alta complexidade, será estabelecida em norma editada pela ANS, que publicará rol de procedimentos e eventos em saúde suplementar, atualizado a cada incorporação.
[7] BALDWIN, Robert. Understanding regulation. 2a edição. Oxford, P.106
[8] Art. 2º, inc. I.
[9] FREEMAN, Jody. Agency Coordination in shared regulatory space. Havard Law Review. Vol 125. Nº5. Março 2012
[10] Idem. P. 1145. Em tradução livre: atribuições jurisdicionais relacionadas, nas quais o Congresso atribui funções intimamente relacionadas, mas distintas, a várias agências em um regime regulatório ou administrativo mais amplo.
[11] MARISAM, Jason. Duplicative Delegations. Administrative Law Review. Vol 63, nº 2, 2011, P. 215
[12] BORGES, Jose Francisco. O princípio da autonomia da vontade como garantia da moralidade em Kant. Dissertação de mestrado em filosofia. Universidade Federal de Santa Maria. 2007
[13] CAMPOS, Adriana. A relação entre o princípio da autonomia e o princípio da beneficência (e não-malificência) na bioética médica. Revista Brasileira de Estudos Políticos | Belo Horizonte | n. 115 | pp. 13-45 | jul./dez. 2017
[14] HOSSNE, William Saad, Dos referencias da bioética – a vulnerabilidade. rev. Bioethikos. 2009;3(1):41-51
[15] CERVI, Taiana Damo. Cuidados paliativos e autonomia do paciente terminal: reflexões sobre o testamento vital no Brasil. Revista Videre, Dourados, MS, v.10, n.20, jul./dez. 2018